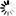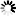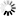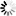S-亚硝基化修饰 EZH2:调控内皮稳态的新机制与潜在治疗靶点
摘要:研究人员开展了关于S-亚硝基化修饰EZH2的研究。
在生命的微观世界里,细胞的正常运作依赖于各种精细的调控机制。一氧化氮(NO)作为一种多功能的生物活性分子,在细胞功能调节中扮演着重要角色,它可通过S-亚硝基化修饰蛋白质来发挥作用。EZH2是多梳抑制复合物2(PRC2)的关键功能性酶组分,负责催化组蛋白H3第27位赖氨酸的甲基化(H3K27me3),进而调控基因表达,在细胞分化、发育以及多种疾病进程中都有着举足轻重的地位。此前,虽然NO信号通路对血管内皮细胞基因表达有影响,但NO依赖的蛋白质S-亚硝基化修饰在定义内皮细胞表观遗传景观中的作用尚不明确,EZH2的S-亚硝基化修饰及其对该关键表观遗传调节因子的催化活性、定位和降解的影响也未见报道。为了填补这些知识空白,来自印度比拉理工学院(Birla Institute of Technology and Science)的研究人员开展了深入研究,相关成果发表在《Nature Communications》上,为理解内皮细胞功能调控及相关疾病治疗提供了新的视角。
研究人员主要运用了以下几种关键技术方法:一是使用生物素开关法(biotin switch assay)和碘代串联质谱标签(iodoTMT)标记试剂检测EZH2的S-亚硝基化;二是通过免疫共沉淀(co-immunoprecipitation)实验探究蛋白质之间的相互作用;三是利用染色质免疫沉淀(Chromatin Immunoprecipitation,ChIP)及后续的定量PCR分析基因启动子区域H3K27me3的富集情况;四是借助分子动力学(MD)模拟从结构层面分析S-亚硝基化对EZH2-SUZ12复合物的影响。
 图1EZH2的S-亚硝基化通过改变PRC2复合物的组装、甲基转移酶活性及EZH2的稳定性来维持内皮稳态
图1EZH2的S-亚硝基化通过改变PRC2复合物的组装、甲基转移酶活性及EZH2的稳定性来维持内皮稳态SNP/GSNO与EZH2的相互作用及对相关指标的影响
研究人员以硝普钠(SNP)作为外源性NO供体,发现SNP处理人脐静脉内皮细胞(HUVEC)和人脐静脉内皮细胞系(EA.hy926细胞)后,细胞内亚硝酸盐水平呈时间依赖性增加。SNP可使EZH2蛋白及其催化产物H3K27me3水平降低,且H3K27me3水平在SNP处理1小时后就显著下降,早于EZH2蛋白在2小时后的降解。进一步研究表明,SNP处理30分钟时,SUZ12就从EZH2结合的PRC2复合物上解离,导致PRC2复合物催化活性降低。同时,SNP促使EZH2发生胞质转位,通过亚细胞分级分离和免疫荧光实验得以证实。为排除SNP作用的非特异性,研究人员使用S-亚硝基谷胱甘肽(GSNO)进行实验,得到了相似的结果,且GSNO处理后EZH2的相互作用蛋白发生改变,通过质谱分析鉴定出了相关变化的蛋白。
EZH2的S-亚硝基化修饰及对PRC2复合物活性的影响
研究人员通过体外无细胞系统和细胞实验证实,NO处理可使EZH2蛋白发生S-亚硝基化修饰。生物素开关法和免疫沉淀实验表明,在EA.hy926细胞中,NO处理后EZH2蛋白被S-亚硝基化。同时,使用天然诱导剂缓激肽(bradykinin)诱导内源性NO产生,也检测到EZH2蛋白的S-亚硝基化及与SUZ12结合的丧失。此外,H3K27me3组蛋白甲基转移酶(HMT)活性测定显示,SNP或GSNO处理后,PRC2复合物的甲基转移酶活性显著降低,说明EZH2的S-亚硝基化导致SUZ12从PRC2复合物上早期解离,进而降低其催化活性。

图2一氧化氮通过诱导SUZ12解离,引发EZH2蛋白的胞质定位与降解,并伴随H3K27me3水平的早期下降
EZH2降解途径及内源性NO抑制的影响
研究发现,S-亚硝基化的EZH2主要通过自噬溶酶体途径降解。使用蛋白酶体抑制剂MG132不能逆转SNP处理后EZH2的降解,而自噬溶酶体途径抑制剂巴弗洛霉素A1(bafilomycin A1)可部分逆转。同时抑制蛋白酶体和自噬溶酶体途径,能完全逆转EZH2的降解,但无法恢复H3K27me3的水平。此外,抑制内源性NO产生机制,如使用eNOSsiRNA敲低HUVEC细胞中的eNOS,或用L-NAME抑制一氧化氮合酶(NOS),可逆转由VEGF或缓激肽诱导的EZH2和H3K27me3水平的降低,且限制EZH2的胞质转位,使其定位在细胞核中。
EZH2-H3K27me3轴在糖尿病肾病中的作用及调节效果
研究人员发现,在糖尿病肾病中,EZH2-H3K27me3轴增强。预先用H3K27me3特异性去甲基化酶抑制剂GSK-J4处理细胞,可逆转SNP诱导的H3K27me3水平降低,并抑制SNP诱导的内皮细胞迁移。通过实时定量PCR(qPCR)和染色质免疫沉淀实验发现,SNP处理后,与内皮功能相关的基因如VEGFa、TBX20等的转录水平增加,且这些基因启动子区域H3K27me3的富集减少。在高糖环境下,GSNO处理可逆转高糖诱导的EC和大鼠主动脉中EZH2、H3K27me3水平的升高以及炎症黏附分子ICAM1的表达,减少单核细胞黏附,这表明S-亚硝基化介导的EZH2调节可能是对抗糖尿病血管并发症的潜在治疗策略。
EZH2特定半胱氨酸残基S-亚硝基化的作用及分子机制
通过预测工具发现EZH2蛋白的半胱氨酸329(C329)、700(C700)等位点可能发生S-亚硝基化。构建点突变体实验表明,C329S突变体对SNP处理不敏感,其EZH2蛋白水平未受影响,但H3K27me3水平显著降低;C700S突变体的EZH2蛋白水平在SNP处理后显著下降,而H3K27me3水平不变。双突变体EZH2 C329S C700S对SNP完全不敏感。免疫荧光和共聚焦成像显示,SNP可诱导野生型EZH2的胞质转位,但对C329S和双突变体无此作用。分子动力学模拟显示,EZH2在C329和C700位点的S-亚硝基化导致EZH2-SUZ12复合物构象改变,使SUZ12与EZH2的SAL结构域结合减弱,最终导致复合物不稳定。
综上所述,该研究揭示了NO信号通路通过S-亚硝基化修饰EZH2的C329和C700残基,影响PRC2复合物的稳定性和催化活性,进而调控基因表达,维持内皮稳态。在糖尿病等疾病状态下,S-亚硝基化修饰EZH2可能成为潜在的治疗靶点。不过,研究也存在一定局限性,如未能在体内模型中证实eNOS基因表达或催化抑制对EZH2和H3K27me3的影响。未来研究可进一步探索NO依赖的表观遗传通路调控机制,为相关疾病的治疗提供更坚实的理论基础和潜在的治疗策略。
参考资料
[1]S-nitrosylation of EZH2 alters PRC2 assembly, methyltransferase activity, and EZH2 stability to maintain endothelial homeostasis
 |
 |
 |
| 官网:www.cxbio.com | 微信服务号:iseebio | 微博:seebiobiotech |
 |
 |
 |
| 商城:mall.seebio.cn | 微信订阅号:seebiotech | 泉养堂:www.canmedo.com |
相关资讯
- Immunity:我国科学家揭示靶向巨噬细胞中的胆固醇代谢可清除病毒感染
- 大肠杆菌宿主细胞蛋白ELISA试剂盒
- Nature:利用CRISPR,终于弄清楚了一种独特的免疫细胞是如何识别并摧毁肿瘤的
- 实验“搭子”喊话:快来查收各类常用动物造模试剂
- 无荧光性新型SHG成像用染料 - Ap3
- 金属螯合介质(NTA)——层析介质优选西宝生物
- 何时须更换培养基?
- 一种以前未知的机制:当细胞的DNA受损时,这种机制会引发细胞的炎症免疫反应
- FITC标记DEAE葡聚糖
- Phos-tag(TM) 凝胶荧光染料

")

新进产品
同类文章排行
- S-亚硝基化修饰 EZH2:调控内皮稳态的新机制与潜在治疗靶点
- Nature|华大携手瑞典乌普萨拉大学发表全球最大规模结直肠癌多组学研究
- 细胞表面的RNA结合蛋白是对抗癌症的关键
- 抗真菌蛋白DECTIN-1可用于自身免疫疾病和癌症治疗
- Science解开长期谜题:破译血清素在我们大脑中的作用
- 《自然》突破性成果:解决mRNA疫苗的不足之处
- 阿尔茨海默病SIRT2酶与记忆修复的希望
- Nature解决难题:超灵敏游离 RNA(cfRNA)检测方法
- Nature:科学家新发现一种免疫细胞类型,与过敏有关
- Nature:在大脑免疫细胞中发现潜在的阿尔茨海默病治疗靶点
资讯文章
您的浏览历史